

What is hemophilia?
Hemophilia is a genetic disorder that diminishes the body’s ability to make blood clots that are needed to stop bleeding. In hemophilic patients, bleeding continues for a long time after an injury, easy bruising, and an enhanced risk of bleeding within joints or the brain. Mild cases of the condition may have symptoms only after an accident or during surgery.
Bleeding into a joint may result in permanent damage while in the brain may end up in long term headaches, seizures, or lack of awareness.
How does bleeding start and stop?
- Bleeding begins when a capillary is hurt, and blood flows out.
- The capillary contracts to reduce the bleeding.
- Then platelets make a plug to patch the crack.
- Next, numerous clotting factors in plasma cooperate to build up a clot that makes the plug more effective and stops the bleeding.
How does hemophilia affect the blood?
Platelets are essential for blood clotting as they have an adhesive surface that allows them to tramp together to stop the leak of blood.
Clotting factors are proteins that form a “network” around the platelets, supporting them to stay in position.
There are various clotting factors in the blood that enumerated using Roman numbers.
A child with hemophilia A does not have sufficient clotting factor VIII (8) in their blood. A child with hemophilia B does not have adequate clotting factor IX (9) in their blood.
In hemophilia, the missing clotting factor makes it hard to build a clot. Your bleeding will remain longer than usual.
⇒ Now, let’s discuss types of hemophilia.
Types of hemophilia


1) (Classic Hemophilia): This type occurs due to low amounts of clotting factor VIII.
2) Hemophilia B (Christmas Disease): This type occurs due to low levels of clotting factor IX.
3) Hemophilia C: This type occurs due to low levels of factor XI.
4) Para hemophilia: This type occurs due to low levels of factor V.
5) Acquired hemophilia: This type of hemophilia occurs with cancers, autoimmune disorders, and pregnancy.
Most of the affected children inherit hemophilia from their parents, who carry an abnormal gene on the X chromosome. But, an unusual mutation may occur later in life, or an autoimmune disease attacks the clotting factors causing hemophilia.
Who is Affected?


Hemophilia A is about four times as frequent as hemophilia B.
As hemophilia A and B are both X-linked recessive disorders, it is popular in males, and females are unusually affected.
Some women with an abnormal gene on one of the X chromosomes may be kindly symptomatic.
Hemophilia C occurs equally in both sexes and mostly in Ashkenazi Jews.
In the 1800s, hemophilia B was well-known within the royal families of Europe.
› Diagnosis depends on the analysis of the blood capability to clot and the levels of clotting factors.
› Prevention may occur by separating an egg, fertilizing it, and examining the embryo before transferring it to the uterus.
› Treatment is by substituting the absent blood clotting factors. We do that regularly or during bleeding episodes. We can do this replacement at home or in the hospital.
Hemophilia A
Hemophilia A is a hereditary disorder caused by defective clotting factor VIII. Other names are factor VIII (FVIII) deficiency or classic hemophilia
Although hemophilia is a genetic disorder passed down from parents to children, about 1/3 of cases appear due to a spontaneous mutation.
Hemophilia B


Hemophilia B, also titled factor IX deficiency or Christmas disease, is the second most popular type of hemophilia. The disorder was first announced in the medical history in 1952 in a patient with the name of Stephen Christmas. The most influential family with hemophilia B was that of Queen Victoria of England.
Acquired hemophilia B occurs when the body produces antibodies against its factor IX protein. The factor IX antibodies suppress circulating factor IX in the blood, resulting in bleeding symptoms. Acquired hemophilia B is a rare condition.
Hemophilia a and hemophilia b
| Hemophilia A | Hemophilia B |
| low levels of factor VIII (8) | low levels of factor IX (9) |
| treated with factor VIII replacement | Treated with factor IX replacement |
| Prophylaxis:
hemophilia A needs preventative injections three times per week or every other day. |
Prophylaxis:
Severe hemophilia B needs preventive injections twice a week. |
Hemophilia A and B are congenital bleeding disorders caused by a low level or lack in one of the factors usually found in the blood that make a stable blood clot.
A sick blood clot puts individuals at risk of bleeding inside the joints or muscles. Bleeding can happen when moving ordinarily or exercising or straining, or due to an injury, and a clot will take longer to develop and stop the bleeding. The individuals may re-bleed when the sick blood clot breaks down quickly.
People with zero levels of the factor have severe hemophilia, and these people are at higher risk of bleeding. People with higher levels up to the normal ranges have moderate or mild hemophilia, and the danger of bleeding is less for them.
Hemophilia C

Hemophilia C affects one person in every 100000. But in Ashkenazi Jews, this type of hemophilia affects 8% of the population due to intermarriage. Hemophilia C is an autosomal recessive disorder, which requires an abnormal gene in both parents to occur in their children. Hemophilia C affects both sexes at an equal rate. The role of factor XI is vital in the coagulation process because it activates more thrombin, which converts fibrinogen into fibrin. Fibrin aggregates mare platelets and makes the clot harder.
Symptoms don’t resemble with Factor XI levels in the blood. Individuals with lower levels may bleed less than those with higher levels of Factor XI. Patients may undergo nose bleeds or soft tissue bleeds, as well as hemorrhaging after tooth removal.
Many women may not recognize the deficiency in factor XI until they suffer from menorrhagia (heavy menstrual periods) or postpartum bleeding.
In hemophilia C, joint and muscle may bleed, but this condition is uncommon.
Doctors will command a bleeding time test, platelet function tests, and prothrombin time (PT)tests.
Doctors need activated partial thromboplastin time (aPTT) tests to confirm the diagnosis of hemophilia C.
Factor XI concentrates are unavailable, so doctors manage hemophilia C with fresh frozen plasma.
Fibrin glue is suitable to maintain clots and control mouth bleeds. When mixed with fresh frozen plasma, it arrests bleeding following circumcision and hernia adjustment.
Antifibrinolytics can control nose bleeds or bleed after tooth extraction.
Hemophilia causes


Hemophilia occurs due to a problem in one of the genes that tell the body to produce the clotting factor proteins needed to form a blood clot.
Even though hemophilia is genetic, it may occur among families with no previous history. About one-third of recently diagnosed kids have no family history of hemophilia. These conditions are due to gene mutation (a change to the gene’s directions for building up the clotting factor protein). Gene mutation can prevent the clotting protein from acting accurately or prevent its multiplication.
Acquired hemophilia
Acquired hemophilia is an autoimmune disorder where the immune system attacks blood clotting factor VIII and represses its role, leading to excessive bleeding occurs in patients with a personal and family history negative for hemorrhages.
The disease influences males and females equally, but it usually develops later in life.
About 50% of the cases of acquired hemophilia are idiopathic (unknown cause). Acquired hemophilia may occur with autoimmune diseases, such as rheumatoid arthritis, psoriasis, or ulcerative colitis. Rarely, cancer or some drugs may be the cause.
Pregnant females may have acquired hemophilia during the first year after pregnancy, and this represents the majority of acquired cases in females younger than 40 years old.
» We have two chief types of acquired hemophilia :
1. acquired hemophilia A is due to factor VIII deficiency.
2. acquired hemophilia B is due to factor IX deficiency.
But, acquired hemophilia may involve other clotting factors in limited conditions.
Specific symptoms


The most common symptom of acquired hemophilia is ecchymosis, which means subcutaneous bleeding. Also, the blood may collect in the muscles forming am mass called a hematoma. Bleeding may occur from the gastrointestinal tract (melena), the genitourinary tract (hematuria), or retroperitoneal. Bleeding per nose (epistaxis) and bruises (all over the body) may occur.
Pain may occur in the arms and legs because the bleeding may compress the muscles, nerves, or blood vessels.
Patients may bleed during surgical operations or after minor trauma. Pregnant women may have genital bleeding, especially after delivery.
Intracranial hemorrhage rarely occurs, but it is fatal.
In contrast to congenital hemophilia A,
- Joint bleeding is infrequent.
- Bleeding occurs without cause (spontaneously).
- Bleeding episodes are severe and may become life-threatening.
- Bleeding into the soft tissues may progress rapidly, causing a compartmental syndrome.
- If left untreated, acquired hemophilia may be life-threatening.
› causes
1) Autoimmune
The immune system reacts to a foreign substance by producing specific proteins called antibodies. Antibodies act by crushing foreign substances directly or by coating them with a material that distinguishes them for crushing by white blood cells.
Autoantibodies are antibodies that target healthy tissues. In acquired hemophilia, these antibodies attack the clotting factor and inhibit it.
2) Idiopathic
No underlying disorder or triggering crisis can be recognized.
3) Pregnancy
Association with pregnancy is announced mostly in the Postpartum Period.
4) Coexisting disorder, such as lupus, rheumatoid arthritis, multiple sclerosis, inflammatory bowel disease, cancer, diabetes, or hepatitis
› Affected Populations
AH develops in
1) individuals with no history of bleeding disorder with approximately equal fractions of males and females affected.
2) Individuals of any age can be affected, although AH is infrequent in children.
3) The occurrence increases with age and affects old individuals (between 60-80 years of age).
4) A small increase in incidence occurs in pregnant between the ages of 20_40.
› Diagnosis
Patients present prolonged bleeding, or extensive bruising on the trunk, legs, and arms, that appear distinct from usual injury-related bruising.
We confirm the diagnosis by an abnormal coagulation test that shows an isolated prolongation of the activated partial thromboplastin time (aPTT).
A simple blood analysis can discover if there a lack of clotting factor VIII.
Immunosuppressive drugs and injections of clotting factors are excellent items for treatment.
› Treatment
Treatment will vary depending on the specific symptoms present. Therapeutic tactics depend on control bleeding (if present or significant), elimination of the inhibitor, and treatment of the related disease (if applicable).
› Control Bleeding Episodes
Doctors should start anti-hemorrhagic treatment in AH cases, irrespective of inhibitor level, and factor VIII activity.
- use of bypassing agents (bolus of factors that restore the acquired deficiency)
- Bypassing agents are the favored first-line treatment due to their fast response and high level of effectiveness.
- increase FVIII levels.
- Fibrin glue or antifibrinolytic agents may control local bleeding efficiently.
Hemophilia genotypes and genetics


» Hemophilia A and B are called X-linked recessive conditions, indicating the gene that induces hemophilia is on the X chromosome.
Males own one X chromosome (XY), and females have two X chromosomes (XX). Therefore, hemophilia usually affects males. When a male has received a gene that induces hemophilia on his X chromosome, he does not provide the clotting factor he requires because the Y chromosome does not store information for the production of factor VIII or IX.
When a female receives a gene that induces Hemophilia on one of her X chromosomes, she owns a second normal X chromosome. Therefore, the healthy gene produces the essential clotting factors and compensates for the hemophilia gene.
These women will be carriers of hemophilia.
» Men with hemophilia pass on hemophilia gene to all of their daughters, so they will be carriers.
But, their sons don’t receive the abnormal gene as they receive Y, not X chromosome, so they will not have hemophilia.
» Women who are carriers have a 50% opportunity to transfer the hemophilia gene to their kids.
Each son of a carrier has a 50% opportunity to have hemophilia, and each daughter has a 50% opportunity to be a carrier.
If a carrier woman is pregnant (gender unknown), she has a 25% opportunity to have a son with hemophilia, and a daughter who is a carrier. She may have a son or daughter who does not have hemophilia and is not a carrier with a 50% opportunity. If her first son is born without hemophilia, the subsequent male may have hemophilia with a 50% chance. The chance to transfer the hemophilia gene is the same with each pregnancy, regardless of her previous offspring conditions.
» Women who are carriers may suffer bleeding manifestations, as they may have low factor levels sufficient to be in the mild range of deficiency.
Females who carry hemophilia should have their factor (VIII or IX) levels monitored to ensure they have sufficient levels for natural blood clotting.
» If you are a carrier, women in your family may carry the gene for hemophilia. Therefore, there may be other family members at risk.
» Genetic examination is accessible for individuals with hemophilia and women at risk of being carriers.
» We should encourage our patients to make their family aware of hemophilia items. Knowledgable individuals take care of themselves and obtain proper treatment and information.
How is hemophilia inherited?


The abnormal gene is on the X chromosome. It can be carried by either the mom or dad or both.
The opportunity of a baby receiving a hemophilia mutated gene depends on which of their parents has the mutated gene.
If only the mother has the mutated gene
If a female with the mutated gene and a normal man has a kid, there is a:
- 1 in 4 opportunities of having a normal baby boy
- 1 in 4 chance of having a baby boy with hemophilia
- 1 in 4 chance of having a normal baby girl
- 1 in 4 chance of having a baby girl with an affected X chromosome
In the last state, the girl becomes a carrier of the mutated gene. The carrier girl may transfer it to her kids but may not have any severe manifestations of hemophilia herself.
However, female carriers may have bleeding problems, such as heavy.
If only the father has the changed gene
If a male with hemophilia has a son with a normal woman, there’s no opportunity the son will get hemophilia.
Because the boy always receives his X chromosome from his mother, who does not have the mutated gene.
However, any girls of the man will become carriers of the mutated hemophilia gene and may transfer it to their kids.
If both parents have the mutated gene
If a female with the mutated gene and a male with hemophilia have a kid, there’s a:
- 1 in 4 opportunities of having a normal baby boy
- 1 in 4 chance of having a baby boy with hemophilia
- 1 in 4 chance of having a baby girl who’s a carrier of hemophilia
- 1 in 4 chance of having a baby girl with hemophilia
So, females may have hemophilia, although it’s very uncommon.
When there’s no family history
A boy may be born with hemophilia, although there’s no family history of the disorder.
In such events, the gene mutation developed spontaneously, in the son’s mother, grandmother, or great-grandmother, but till then, a son of the family had never received it.
Some studies have shown, there’s no recognized family history of hemophilia in up to 1 in 3 new cases.
Genetic mutation
A human can acquire all genetic disorders spontaneously by mutation, rather than inheriting it, because of a new mutation in one of their parents’ gametes. Spontaneous mutations estimate about 33% of all cases of hemophilia A.
About 30% of states of hemophilia B are the outcome of a spontaneous gene mutation.
So, females may deliver a hemophiliac son, she is a carrier for the disorder or not, as a result of a spontaneous mutation.
Until modern direct DNA trial, however, it was difficult to determine if a female with only healthy kids was a carrier or not. Generally, the healthier sons she bore, the higher the probability that she was not a carrier.
Is hemophilia dominant or recessive
Hemophilia is an X-linked recessive condition.
It is a recessive disorder because the normal X chromosome dominates the diseased one in a carrier woman.
In the affected male, the mutated gene acts alone and causes the disorder.
Hemophilia pedigree


Pedigree is a method that describes the inheritance of the inherited disease, and the previous pedigree describes the pattern of hemophilia in Queen Victoria’s children. Question marks indicate the kids whose case is unknown. The descendants in the third and fourth generations didn’t receive the mutant gene; thus, we deleted them.
New work explains that the mutation was in the gene for factor 9 (IX), so the condition was hemophilia B. The examination of the DNA samples of the Russian Royal family showed:
- A point mutation in factor IX (F9) gene
- The mutation site caused a frameshift that resulted in a shortened, nonfunctional protein.
- As the pedigree shows, the DNA of Alexandra contained a mutant allele; thus, she was a carrier.
- Was this one of her four Daughters (referred to as “4?” in the fourth row and Presumably Anastasia)?
- However, the DNA of Czarevitch Alexis contained only the mutant allele, which accounts for his previous history of severe bleeding.
Signs and symptoms of hemophilia


The symptoms of hemophilia depend on the severity of the condition, but the chief indication is prolonged bleeding.
The bleeding may occur spontaneously, so we observe
- sudden nosebleeds
- bleeding gums
- bleeding within joints and muscles
The bleeding may occur after a medical or dental intervention, such as a tooth extraction.
A) Mild hemophilia
Kids born with mild hemophilia may not have any signs for years.
Mild hemophilia usually becomes manifest after a wound, or surgery, or a dental procedure such as having a tooth removed.
B) Moderate hemophilia
Kids with moderate hemophilia are affected like those with mild hemophilia, but they bruise easily.
They may have manifestations of internal bleeding around their joints, especially if they have a blow or a fall that hits their joints (joint bleed).
The manifestations usually start with a tingling feeling of irritability and mild pain in the involved joint, especially the ankles, knees, and elbows. Shoulder, wrist, and hip joints can be affected.
If we ignore joint bleed, it can lead to:
- more severe joint pain
- stiffness
- the bleeding area becomes warm, swollen, and tender
C) Severe hemophilia
The manifestations of severe hemophilia are related to the moderate form of hemophilia. However, joint bleeding is more severe.
Kids with severe hemophilia have spontaneous bleeding.
Bleeding starts without a definite cause and may develop:
- nosebleeds
- bleeding gums
- joint bleeds
- muscle bleeds
Without adequate therapy, individuals with severe hemophilia may develop:
- joint deformity, which may need joint replacement surgery
- soft tissue bleeding
- severe internal bleeding
Bleeding inside the skull, identified as a brain or subarachnoid hemorrhage.
However, spontaneous bleeding inside the skull is rare and induced by a head injury.
Bleeding in the skull is a life-threatening condition that requires urgent intervention.
The symptoms of a brain hemorrhage include:
- neck stiffness
- severe headache
- projectile vomiting
- blurring vision
- an altered mental state such as confusion
- difficulty speaking, such as slurred speech
- loss of coordination of the facial muscles
Females carriers produce adequate clotting factors from their one normal gene helping them to limit bleeding disorder, but some may manifest as mild hemophilia manifestations.
Complication
Complications are frequent with severe and moderate hemophilia and may result from the disease itself or its therapy:
- Internal bleeding leads to swelling, numbness, or pain of a limb.
- Joint damage from hemarthrosis (hemophilic arthropathy) leads to a painful and deformed joint. Debilitating arthritis may develop.
- Infections occur as a result of using blood transfusions as treatment.
- Intracranial hemorrhage is a life-threatening condition caused by the increase of pressure inside the skull. It can create a loss of consciousness, brain damage, and even death.
- Adverse reactions to clotting factor treatment include the improvement of an immune inhibitor, which opposes factor replacement intervention.
Hemophilic arthropathy is a mix of chronic proliferative synovitis and cartilage damage. The untreated intra-articular bleed may cause apoptosis of chondrocytes and influence the construction of proteoglycans. Iron deposition in the synovium may induce an inflammatory response stimulating the immune system and stimulating angiogenesis, followed by cartilage and bone destruction.
Hemophilia bruises
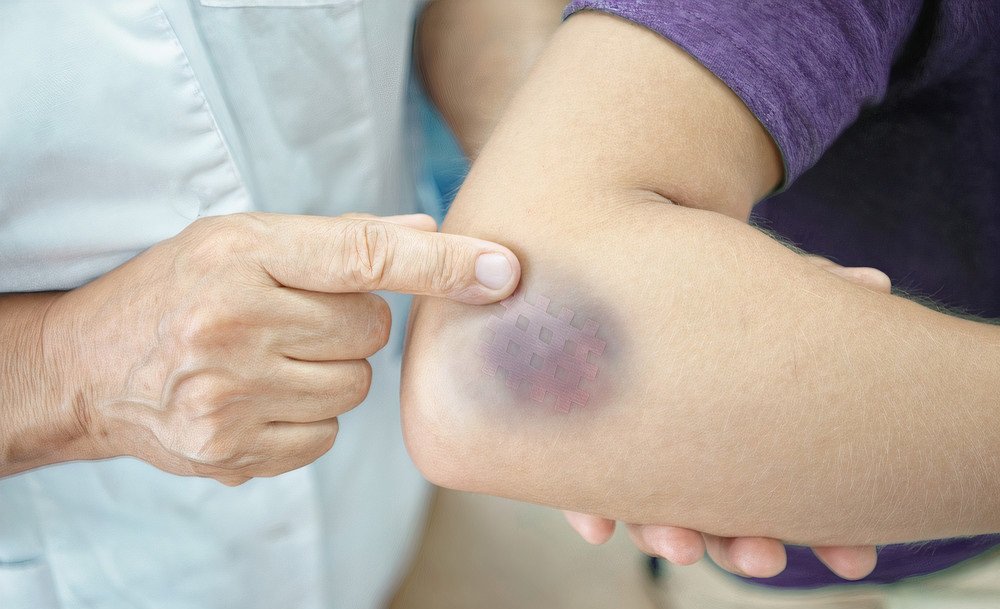

A bruise, or contusion, is a skin discoloration due to tissue injury that damages blood vessels beneath the skin, causing them to leak. When blood flows beneath the skin, it causes black, blue, purple, brown, or yellow discoloration.
When the skin breaks open, external bleeding occurs.
Hematomas are painful, enlarged bruises that can influence internal organs. Hematomas may require medical awareness.
Who might get a bruise?
Bruises can happen from an accident, sports injury, or medical intervention. Old individuals can bruise easily. Bleeding disorders, such as Hemophilia, can lead to excessive bruising.
You may be more exposed to bruising in the following situations:
- Cancer
- Liver diseases
- Positive family history of bruises
- Nonsteroidal anti-inflammatory drugs (NSAIDs) for pain relief, such as ibuprofen or naproxen
- Medications that alter the blood coagulation process, such as aspirin or blood thinners
- Hemophilia and von Willebrand disease
- Low blood platelet count (thrombocytopenia)
- Deficient vitamin C or vitamin K
Types of bruises include:
- Hematoma: It is an accumulation of blood outside the blood vessels that produces pain and swelling.
- Purpura: It is small bleeding that occurs under the skin.
- Petechiae mean red spots on the skin that don’t become white after a gentle pressure.
- Senile purpura: Type of purpura occur in elder individuals as their skin becomes thinner and dryer, which makes it more susceptible to tearing.
- Black eye: It is a discolored ring around the eye, which occurs due to a head blow.
It may indicate a severe eye injury, such as bleeding in the eye (hyphema), or a facial fracture.
Signs of bruising:
They may look red or purplish at first, but with darker skin, you will notice purple, dark brown, or black bruising. During healing, the bruise may shift a brighter brown, green, or yellow.
The bruised region and enveloped skin may be tender to touch.
A hematoma causes a swollen, raised, painful swelling.
Diagnosis of hemophilia


We can diagnose Hemophilia before, during, or after birth if there is a family history of the condition.
Many choices are accessible to parents.
Mild hemophilia may be discovered later, usually after an injury or a dental or surgical intervention.
› Before pregnancy
Genetic measurement and counseling are accessible to determine the risk of transferring the disorder to a child. We examine a sample of tissue or blood to inspect marks of the genetic mutation that induces hemophilia.
› During pregnancy
A pregnant with a family history of hemophilia can examine for the hemophilia gene.
Specific examinations include
- Chorionic villus sampling (CVS): We take part of the placenta from the uterus and examine it for the hemophilia gene. We do that during weeks 11–14 of pregnancy.
- Amniocentesis: We examine a sample of amniotic fluid during weeks 15–20 of pregnancy.
We may have problems while taking samples, such as miscarriage or premature labor.
› After birth
If we suspect hemophilia in a newly born child, we can apply
a blood test to confirm the diagnosis. We can examine blood from the umbilical cord at birth if there’s a family history of hemophilia. We can identify the type of whether a hemophilia A or B and how severe it is by a blood test.
We use umbilical cord blood testing to recognize levels of factor VIII (8), not factor IX (9). Factor IX (9) takes a longer time to develop and reach a standard level at least Six Months of age. Therefore, a low level of factor IX (9) at birth does not mean that the baby has Hemophilia B.
» Hemophilia with no family history,
We can diagnose it when a child starts to walk or drag.
About one-third of babies with hemophilia have no family history.
A doctor might examine for hemophilia in a newborn if:
- Prolonged bleeding after the circumcision of the penis occurs.
- Bleeding in the scalp or brain after hard labor using instruments to deliver the baby happens.
- Large numbers of bruises appear when the child begins standing or dragging.
We can diagnose individuals with severe hemophilia during the first year of life as they suffer severe bleeding problems. We can diagnose people with milder forms of hemophilia until later in life.
Screening tests are blood tests that determine the blood clotting state and help to diagnose.
Types of screening tests:
- Complete Blood Count (CBC) usually shows natural results in patients with hemophilia. But in heavy bleeding, the hemoglobin and the red blood cell count may decrease.
- Activated Partial Thromboplastin Time (APTT): It is a test that measures the activity of factors VIII (8), IX (9), XI (11), and XII (12). The results of this test explain the longer coagulation time in patients with hemophilia A or B.
- Prothrombin Time (PT) Test:It is a test that measures the activity of factors I (1), II (2), V (5), VII (7), and X (10). The results of this test will be within the normal range in patients with hemophilia A and B.
- Fibrinogen Testmeasures the activity of fibrinogen (Factor I)
Clotting Factor Tests diagnose bleeding disorders blood, which shows the type of hemophilia and the severity of the condition, which helps to create the best therapeutic plan.
Severity
The severity of hemophilia differs according to the degree of gene mutations, which affects the factor activity.
Hemophilia


The best treatment of hemophilia is to replace the defective clotting factor, which enables the blood to clot in a natural way.
In mild hemophilia, we don’t require those factors.
In moderate hemophilia, we need clotting factors when bleeding occurs. In severe hemophilia, Doctors recommend preventive use of those factors two or three times per week and may continue for life. Rapid treatment of bleeding episodes decreases harm to the body.
- Hemophilia A requires Factor VIII.
- Hemophilia B requires factor IX.
Factor replacement can be either separated from human plasma, recombinant, or a mixture of the two.
Inhibitors
Part of individuals with hemophilia develops an antibody (called an inhibitor) that inhibits the effect of the clotting factors used to treat bleeding, so treatment becomes remarkably complex. The value of medical care grows because more clotting factor or a different type is needed.
Patients with inhibitors suffer from joint disease and other complications of bleeding that diminish the quality of life.
Inhibitors develop because of the reaction between the immune system with the infused clotting factor.
The immune system marks the factor as a foreign element and provides inhibitors, or antibodies, to destroy the factor. Most inhibitors originate in the initial 75 clotting factor dosage with the highest risk in the first 10-20 dosages.
Inhibitors are most prevalent in patients with severe Hemophilia A.
Other medication
We can use the following treatment:
- Desmopressin (DDAVP) with mild hemophilia A
- Tranexamic acid or epsilon aminocaproic acid prevents the breakdown of clots.
- Pain reliever drugs, steroids, and physical therapy reduce pain and swelling in an affected joint.
- Omalizumab with severe hemophilia A
In acquired hemophilia,
- The most effective treatment is corticosteroids, which eliminate the auto-antibodies in half of the individuals.
- We may use cyclophosphamide and cyclosporine in patients who did not respond to the steroid treatments.
- We may apply high doses of intravenous immunoglobulin or immunosorbent that control bleeding.
Contraindications
- Anticoagulants, such as heparin and warfarin, can worsen clotting difficulties.
- We shouldn’t use drugs that have “blood thinning” side effects, such as drugs that contain aspirin, ibuprofen, or naproxen sodium, because they produce the side effect of prolonged bleeding.
- Patients should avoid activities and sports with a high incidence of trauma, such as motorcycling, boxing, and wrestling.
Hemophilia cure


At this time, there is no cure for hemophilia, but waiting for the results of gene therapy techniques.
Gene therapy for hemophilia is still under experiments. The idea of gene therapy is to use genes or genetic elements to manage or prevent disease. In general, gene therapy involves taking healthy genes (the ones that direct the body to produce clotting factor) and inserting them into the body of a patient with hemophilia. The new genes should increase the clotting factor level in the blood. Successful gene therapy eliminates the need for factor replacement. It cures hemophilia, but the gene may provide a partial cure. Gene therapy may reduce symptoms of severe hemophilia to a mild or moderate form. It is not currently an accepted therapy for hemophilia until now.
Liver transplants may help a small number of people with hemophilia to cure because the liver makes the clotting factor. The new livers they received made adequate amounts of the factor for them. Transplants are extremely dangerous to use as a cure for hemophilia. We do liver transplants only to save someone’s life when the liver has stopped working.
Hemophilia facts


- Hemophilia is an inherited bleeding disorder.
- Hemophilic patients suffer from prolonged bleeding as the blood does not clot accurately.
- We have three main types of hemophilia A, B, and C.
- The types define the deficient clotting factor.
- The clotting factors included are VIII (8) for A, IX (9) for B, and XII (12) for hemophilia C.
- People living with hemophilia A and B require clotting factor according to their needs.
- Hemophilia C is less dangerous than A and B, and bleeds occur following a surgical or dental intervention. People with hemophilia C do not need regular clotting factor intervention.
- Hemophilia A and B are X-linked disorder so affects boys more than girls, but girls can be carriers of the disease. Hemophilia C affects males and females equally.
- Hemophilia A is the most prevalent type affecting one in 5,000 boys, and hemophilia B affects one in 25,000 boys. Hemophilia C affects one in 100,000.
- Doctors discover the complicated cases within a month and the mild cases in the first 18 months.
- There is no cure for hemophilia, but with preventative treatment, people can live healthy lives.